Computational tools
Here is a list of computational tools generated in my group. Our overall aim is to allow the community to explore data under the FAIR (Findable, Accessible, Interoperable, and Reusable) principles, analyse data, visualise it, and obtain better results. The tools range from assembly improvement (older tools), annotation of genomes (Companion), processing scRNA-Seq or visualising data (ParaCell). For people interested in learning about the scRNA-Seq pipeline, have a look at Scampi. If you are interested in collaborating on tools or do a project with us, please don't hesitate us - however, the more precise your inquiry, the more likely we will reply.
First are webservers, then standalone tools and packages and finally older ones.
Selection of Webservers
The Companion Webserver - Annotate your Genomes!
Annotate your fungi, vector or parasite genome, all automated, reference-based. With thousands job per year, a reliable friend to annotate your genomes! We (Sascha Steinbiss and I) built a companion during my time at the Wellcome Sanger Institute to not only annotate genomes, but also do the primary analysis, such as phylogenetic trees, synthesis and orthologs. At Glasgow, William Haese-Hill took over the project, made it more robust/faster and extended the reference base from parasites to vectors and fungi.
Papers:
Annotation and visualisation of parasite, fungi and arthropod genomes with Companion in Nucleic Acids Research. 2024
Haese-Hill W, Crouch K, Otto TD.
Companion: a web server for annotation and analysis of parasite genomes in Nucleic Acids Research. 2016.
Steinbiss S, Silva-Franco F, Brunk B, Foth B, Hertz-Fowler C, Berriman M, Otto TD.

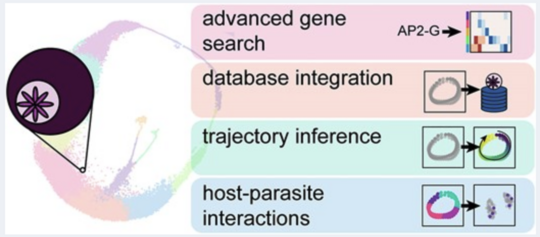
ParaCell - visualise scRNA-Seq data
Visualisation and interpretation of parasite single-cell datasets.
Paper:
paraCell: a novel software tool for the interactive analysis and visualization of standard and dual host-parasite single-cell RNA-seq data. in Nucleic Acids Research 2025
However, while this wealth of available data represents a significant resource, analysing these datasets often requires expert computational skills, preventing a considerable proportion of the parasitology community from meaningfully integrating existing single-cell data into the …
Lead: Edward Agboraw
Exploring Single-Cell Data: Introducing SCAMPI
Analyzing single-cell data is more challenging than traditional methods. Because the data can be noisy, researchers need to carefully adjust settings and make decisions about how much detail to focus on, like figuring out the right number of categories or clusters. This often requires learning new computer programming languages like R or Python.
To make this process easier, we've created SCAMPI, a user-friendly web server. SCAMPI guides you through the entire single-cell analysis process using a popular tool called ScanPy. With SCAMPI, you can:
- Experiment with different settings: See how changing parameters affects your results.
- Learn the steps involved: Understand the different stages of the analysis process.
- Visualize the results: Explore interactive graphs to gain insights from your data.
SCAMPI is designed to be efficient, supporting up to 40 users simultaneously using only 8 CPU cores and 40 GB of memory.
We're currently developing more resources to help even more people explore the power of single-cell analysis.
About SCAMPI:
SCAMPI was initially created by Robert Brewer during an internship in our lab, under the guidance of Andrew McCluskey. Andrew continued to develop SCAMPI, adding new features as part of his PhD research.

Stand alone tools / packages
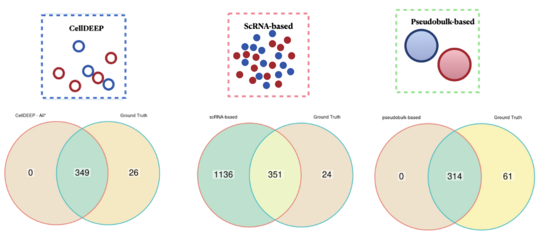
Cell DiffErential Expression by Pooling (CellDEEP) highlights issues in differential gene expression in scRNA-seq
CellDEEP is a tool for analyzing single-cell RNA sequencing data to identify genes that change between conditions. Traditional methods often struggle with noise—either detecting too many false signals or missing important ones.
CellDEEP overcomes this by grouping similar cells together, reducing noise while keeping meaningful biological information. This leads to more reliable results and a clearer understanding of how cells respond in health and disease.
Tested on both simulated and real-world datasets, CellDEEP improves accuracy and helps researchers gain more trustworthy insights from single-cell data.
Lead: Yiyi Cheng, Domenico Somma
Git-hub (soon on Bioconductor)
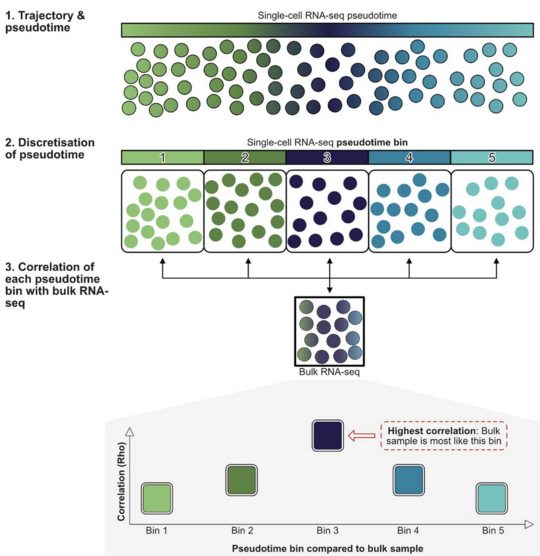
BLASE: Bulk Linkage Analysis for Single Cell Experiments - Teasing Out the Secrets of Bulk Transcriptomics with Trajectory Analysis
BLASE is a computational tool that links bulk RNA sequencing data to dynamic biological processes captured by single-cell data. While bulk RNA-seq is widely used and cost-effective, it cannot easily reveal how cells progress through processes such as development or disease.
BLASE overcomes this by mapping bulk samples onto trajectories learned from single-cell RNA sequencing, allowing researchers to estimate where a sample lies along a biological process.
This enables a better interpretation of existing datasets, supports more accurate comparisons between conditions, and helps uncover changes in disease progression or cellular development.
Github - or via Bioconductor: BiocManager::install("blase")
Lead: Andrew McCluskey
upsML: A high-accuracy machine learning classifier for predicting Plasmodium falciparum var gene upstream groups
upsML is a machine learning tool designed to classify highly variable var genes in Plasmodium falciparum, a key driver of malaria severity and immune evasion. Correctly assigning these genes to functional groups is essential for understanding disease mechanisms, but remains challenging—especially when only partial gene sequences are available.
upsML overcomes this by using advanced machine learning models to accurately classify var genes from both full-length and partial sequences, such as commonly used DBLα tags. Compared to existing methods, it achieves significantly higher accuracy while remaining fast and scalable.
By enabling rapid and reliable analysis of large datasets, upsAI supports research into malaria pathogenesis, immune evasion, and epidemiology, helping to better understand and ultimately combat this major global disease.
Lead: Elcid Aaron Pangilinan

RCE: Entropy-Based Spatial Analysis of Interactions in Context
RCE (Regional Co-occurrence Entropy) is a new method for analyzing how different types of entities—such as cells, species, or buildings—interact within their environment. While many spatial methods focus only on proximity, RCE captures how interactions depend on the surrounding context.
By using an entropy-based approach, RCE identifies where specific combinations of categories occur more (or less) often than expected, revealing meaningful patterns shaped by the environment.
We apply RCE across diverse domains, from uncovering immune cell interactions in Alzheimer’s disease, to studying urban diversity and ecological patterns. This versatility makes RCE a powerful tool for gaining deeper insights into complex spatial systems.
Lead: Adnane Nemri
TrAGEDy: Aligning Cellular Trajectories Across Conditions
TrAGEDy (Trajectory Alignment of Gene Expression Dynamics) is a method for comparing how biological processes unfold across different conditions using single-cell data. Many processes—such as development or disease—do not follow the same path, making them difficult to compare with existing approaches.
TrAGEDy overcomes this by directly aligning independent trajectories, avoiding the need for error-prone data integration. This allows researchers to more accurately identify genes and processes that differ between conditions.
Across both simulated and real datasets, TrAGEDy reveals biologically meaningful differences that are often missed by standard methods, enabling deeper insights into complex and asymmetric cellular dynamics.
TrAGEDy—trajectory alignment of gene expression dynamics - 2015
RF Laidlaw, EM Briggs, KR Matthews, A Madany Mamlouk, R McCulloch, TD Otto
Bioinformatics 41
https://github.com/No2Ross/TrAGEDy
Lead Ross Laidlaw
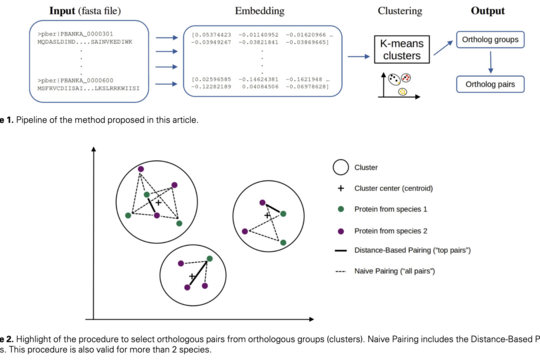
OrthoLM: Discovering Protein Relationships with AI Embeddings
OrthoLM explores how large language models (LLMs) can be used to understand relationships between proteins. Identifying protein homology is key to studying evolution and predicting function, but remains challenging with traditional methods.
By embedding protein sequences using a biologically informed language model and clustering them, OrthoLM can uncover groups of related proteins and identify complex relationships such as orthologs across species.
While complementary to existing approaches, this method demonstrates how AI-driven representations can improve precision and open new possibilities for analyzing protein data at scale.
Minotto T, Claessens A, Otto TD. Bioinformatics. 2025
https://github.com/ThomasGTHB/OrthoLM
Lead Thomas Minotto
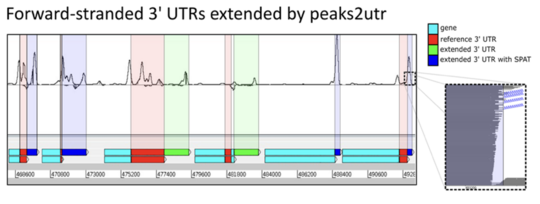
peaks2utr, improving gene annotation with single cell data
eaks2utr is a Python based tool designed to improve gene annotations by identifying 3’ untranslated regions, which are often missing or incomplete, particularly in non model organisms. These regions play an important role in understanding gene expression, yet they are frequently overlooked in standard annotation pipelines.
The tool makes use of single cell sequencing data, such as 10x Chromium, which naturally enriches signals at the ends of genes. By leveraging this information, peaks2utr can accurately extend existing gene annotations and provide a more complete picture of transcript structure.
Easy to use from the command line and freely available, peaks2utr helps researchers generate more reliable annotations, supporting better downstream analysis and interpretation of transcriptomic data.
peaks2utr: a robust Python tool for the annotation of 3' UTRs.
Haese-Hill W, Crouch K, Otto TD. Bioinformatics. 2023
It is available via PyPI at https://pypi.org/project/peaks2utr and GitHub at https://github.com/haessar/peaks2utr.
Lead William Haese-Hill
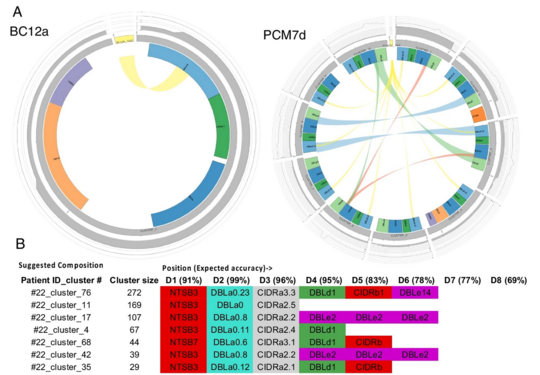
Var gene assemblies, understanding diversity and expression in malaria
These studies focus on the highly variable var gene family in Plasmodium falciparum, which plays a central role in malaria severity by enabling infected cells to evade the immune system and adhere to blood vessels. Due to their extreme diversity, var genes are difficult to assemble and analyse, particularly in clinical samples.
The first study introduces a computational pipeline that improves the assembly and quantification of var gene expression from RNA sequencing data. This approach enables researchers to study var genes alongside the rest of the parasite transcriptome and reveals how gene expression changes when parasites are moved from patients into laboratory culture, highlighting important differences between in vivo and in vitro conditions.
The second study takes a global perspective, assembling var gene repertoires from thousands of parasite isolates across multiple countries. It provides new insights into how these genes evolve, showing both extensive diversity and unexpected sequence sharing across populations, likely driven by recombination and gene flow. Together, these works advance our ability to reconstruct, analyse, and interpret one of the most complex gene families in malaria research.
A novel computational pipeline for var gene expression augments the discovery of changes in the Plasmodium falciparum transcriptome during transition from in vivo to short-term in vitro culture. Andradi-Brown C, Wichers-Misterek JS, von Thien H, Höppner YD, Scholz JAM, Hansson H, Filtenborg Hocke E, Gilberger TW, Duffy MF, Lavstsen T, Baum J, Otto TD, Cunnington AJ, Bachmann A. Elife. 2024
Evolutionary analysis of the most polymorphic gene family in falciparum malaria. Otto TD, Assefa SA, Böhme U, Sanders MJ, Kwiatkowski D; Pf3k consortium; Berriman M, Newbold C. Wellcome Open Res. 2019
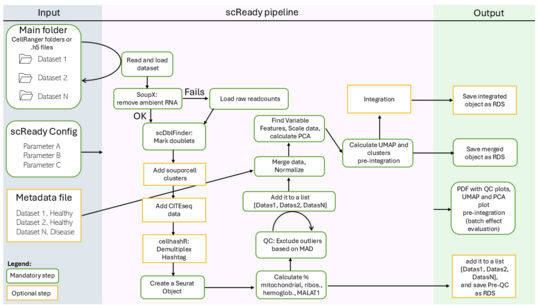
scReady: Simple and Reproducible Preprocessing for Single-Cell Data
scReady is an automated pipeline that simplifies the preprocessing of single-cell RNA sequencing (scRNA-seq) data. While single-cell technologies are powerful, preparing the data for analysis is often complex and requires significant bioinformatics expertise.
scReady streamlines this process by combining all essential quality control and preprocessing steps into a single, easy-to-run workflow. It produces clean, analysis-ready datasets along with clear reports, ensuring reproducibility and transparency.
By lowering the technical barrier, scReady enables researchers,especially those without extensive computational backgrounds, to confidently work with single-cell data and focus on biological insights.
scReady - an automated and accessible pipeline for single-cell RNA-Seq preprocessing: Empowering novice bioinformaticians - Open Wellcome Research
https://github.com/sii-scRNA-Seq/scReady
Lead Domenico Somma
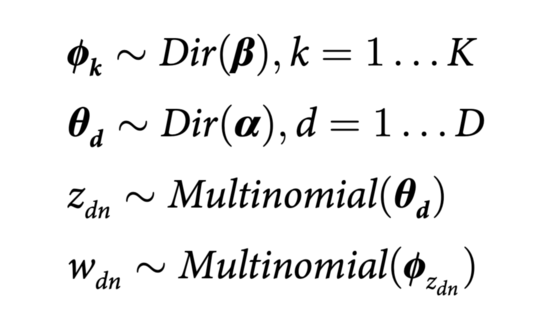
Detecting cellular crosstalk using topic modelling
This work introduces a new computational approach for identifying interactions between cells in single cell RNA sequencing data, without relying on predefined databases of known ligand and receptor pairs. Instead of depending on existing biological knowledge, the method uses topic modelling, specifically Latent Dirichlet Allocation, to detect genes whose expression changes as a result of cell to cell interaction.
The approach takes advantage of recent advances in single cell technologies that allow physically interacting cells to be captured and analysed together. By comparing interacting cells to reference populations, the method can identify genes involved in processes such as adhesion and co stimulation, highlighting both known and previously unrecognised interaction signals.
Tested on both synthetic and real datasets, the method successfully identifies biologically meaningful changes and ranks candidate genes involved in cellular crosstalk. It also works without the need for prior clustering or complex preprocessing steps, making it a flexible and accessible tool for studying cell interactions in a wide range of experimental settings.
Using topic modeling to detect cellular crosstalk in scRNA-seq.
Pancheva A, Wheadon H, Rogers S, Otto TD. PLoS Comput Biol. 2022
Lead Alexndria Pancheva
ILRA: Improving Genome Assemblies from Long Read Data
ILRA is a pipeline designed to improve genome assemblies generated from long read sequencing. While long read technologies have made genome sequencing more accessible, resulting assemblies often remain fragmented and can contain errors, particularly in repetitive regions.
ILRA addresses these challenges by refining assemblies through reordering, merging, and filtering contigs, followed by correction of sequencing errors using short read data. This leads to more complete and accurate genome sequences.
Tested across a range of organisms, ILRA improves assembly quality and reduces annotation errors, helping researchers move from fragmented contigs towards chromosome level genomes.
From contigs towards chromosomes: automatic improvement of long read assemblies (ILRA).
Ruiz JL, Reimering S, Escobar-Prieto JD, Brancucci NMB, Echeverry DF, Abdi AI, Marti M, Gómez-Díaz E, Otto TD. Brief Bioinform. 2023
GitHub: https://github.com/ThomasDOtto/ILRA.
Lead Jose Luiz Ruiz
Not actively supported, but still working
Due to the advances, like long reads, specific tools are not in such high demand. However, they are still be used, like Abacas and RATT in Companion. Please have a look…
Varia, predicting and profiling highly variable malaria genes
Varia is a computational tool designed to analyse highly variable gene families, with a focus on the var genes of Plasmodium falciparum, which encode key proteins involved in malaria virulence and immune evasion. Because these genes are extremely diverse and difficult to reconstruct in full, Varia uses short sequence fragments to predict near full length gene sequences and their domain composition.
The tool includes two complementary approaches. One enables detailed reconstruction of individual genes, supporting experimental validation and structural interpretation, while the other allows rapid profiling of gene expression across complex patient samples. This makes it possible to assess how different variants are expressed and how they may relate to disease processes.
By accurately predicting gene structure and expression patterns from limited data, Varia provides a practical way to study one of the most complex and clinically important gene families in malaria research.
Varia: a tool for prediction, analysis and visualisation of variable genes.
Mackenzie G, Jensen RW, Lavstsen T, Otto TD. BMC Bioinformatics. 2022
Need an update on the blastall, on my to do list
https://github.com/GCJMackenzie/Varia
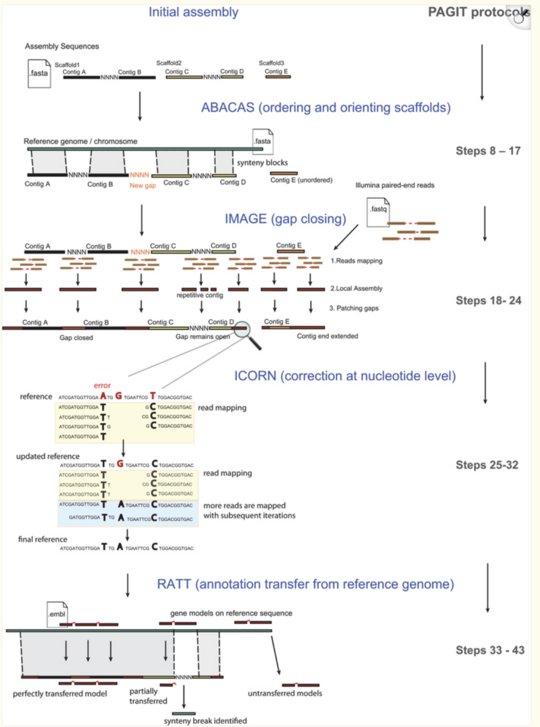
PAGIT: A Toolkit to Improve and Annotate Draft Genomes
PAGIT is a comprehensive toolkit designed to elevate the quality of draft genome assemblies, addressing challenges faced in the modern era of rapid sequencing. While early landmark genome projects involved extensive manual curation, today’s fast output often leaves assemblies less polished. PAGIT provides a flexible pipeline to close gaps, correct errors, and leverage reference genomes for improved scaffolding and annotation. Particularly suited for bacterial and small eukaryotic genomes, PAGIT can significantly enhance assemblies in a short timeframe, as demonstrated with E. coli.
ABACAS is the contig ordering and orientation tool within PAGIT. By aligning contigs to a reference genome with at least 40% amino acid identity, ABACAS arranges and visualizes contigs, and even suggests PCR primers to close gaps if necessary.
IMAGE (Iterative Mapping and Assembly for Gap Elimination) uses paired-end sequencing data to extend contig ends into gaps. By iteratively mapping and assembling reads, IMAGE gradually fills in missing segments, improving the continuity of the assembly.
ICORN (Iterative Correction of Reference Nucleotides) focuses on correcting small errors in the consensus sequence. By repeatedly mapping reads, ICORN fixes single base errors, as well as small insertions and deletions, ensuring a more accurate sequence.
RATT (Rapid Annotation Transfer Tool) facilitates rapid annotation by transferring known gene annotations from a reference genome onto the new draft assembly. Using synteny-based alignment, RATT quickly annotates genes, allowing for functional insights to be gained in a matter of minutes.
Together, these four tools within the PAGIT framework empower researchers to transform raw contigs into annotated, high-quality genomes efficiently.
A post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs.
Swain MT, Tsai IJ, Assefa SA, Newbold C, Berriman M, Otto TD. Nat Protoc. 2012
ABACAS: algorithm-based automatic contiguation of assembled sequences.
Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. Bioinformatics. 2009
– there is an Abacas2 version I would recommedt to use GitHub. It can use multi fasta but just works with nucleotid comparisons
Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology.
Otto TD, Sanders M, Berriman M, Newbold C. Bioinformatics. 2010
RATT: Rapid Annotation Transfer Tool.
Otto TD, Dillon GP, Degrave WS, Berriman M. Nucleic Acids Res. 2011
REAPR: Accurate evaluation of genome assemblies without a reference
REAPR is a versatile tool designed to assess the accuracy of genome assemblies, even when no reference genome is available. Existing methods often rely on qualitative assessments and overlook assembly errors. REAPR addresses these gaps by detecting mis-assemblies and errors with precision. The tool has been validated on various organisms, including bacteria, malaria parasites, and Caenorhabditis elegans. Notably, it revealed that 86% of the human reference genome and 82% of the mouse reference genome are error-free.
When used in ongoing genome projects, REAPR generates accurate assembly statistics, allowing researchers to quantitatively compare different assemblies. In doing so, it helps ensure that genome data is reliable and ready for downstream analyses. REAPR is publicly available from the Wellcome Sanger Institute, providing a valuable resource for the genome research community.
REAPR: a universal tool for genome assembly evaluation.
Hunt M, Kikuchi T, Sanders M, Newbold C, Berriman M, Otto TD. Genome Biol. 2013

RNAcare, linking clinical data with gene expression
RNAcare is an interactive platform designed to help researchers and clinicians explore how gene expression relates to patient specific clinical data. Traditional tools often focus only on RNA sequencing results and require advanced computational skills, making it difficult to connect molecular data with clinical outcomes. RNAcare addresses this by providing an accessible environment that integrates transcriptomic and clinical data in a flexible and user friendly way.
The platform enables users to investigate relationships between gene expression and symptoms such as pain, fatigue, or treatment response, and to identify patterns across patient groups. Applied to rheumatoid arthritis data, RNAcare successfully linked inflammation related genes to clinical symptoms and drug response. It provides a practical tool for generating hypotheses and supporting clinically relevant research.
Tang M, Haese-Hill W, Morton F, Goodyear C, Porter D, Siebert S, Otto TD. BMC Med Genomics. 2025
Old, deprecated, banished - however once nice and valid
For completness here some tools that had a valued purpose.
BAMView: the add on of short read for Artemis my favorite - but not supported - genome viewer. I am still using and will keep an old laptop, until I retire!
ProteinWorldDB: with IBM we compared all existing protein sequences with a Smith-Waterman algorithms, distributed around the world as a screensaver…
AnEnPi: identification and annotation of analogous enzymes, really nice idea… and some of the finding were followed up
ReRep: - finding repetitive sequences, if there was just partial sequencing data available… I wonder if it is possible to have “partial sequencing depth” these days
ChromaPipe: A pipeline to process ABI (Sanger) sequencing data, quick QC, share the data…
During my master 2001!!! I analysed fMRI data with several approaches, SOM, ICA: Model-free functional MRI analysis using cluster-based methods


